Capítulo 16.
DNA recombinante: Las Herramientas del Oficio
Al revelar los numerosos métodos
mediante los cuales las células procesan, añaden, eliminan y transfieren
información genética, los biólogos moleculares abrieron
el camino para el desarrollo de sus propias manipulaciones genéticas.
En los últimos años, se han desarrollado técnicas que han
permitido abordar el análisis y la manipulación del DNA en una
forma antes inimaginada. La tecnología del DNA recombinante ha hecho
posible investigar más a fondo la estructura y función de los
genes, especialmente de los genes eucarióticos que eran inaccesibles
por otros métodos.Estas
investigaciones permanentemente generan nuevos interrogantes e inquietudes,
muchos de los cuales tienen profundas implicancias éticas.
Cuando los investigadores se
enfrentaron por primera vez con el gran tamaño y la complejidad del DNA,
incluso el del virus más simple, la posibilidad de descifrar la información
genética codificada parecía estar más allá de toda
esperanza. Se hizo evidente que para estudiar un gen individual, se lo debía
aislar del resto del genoma ya que, cada gen, representa una pequeña
sección dentro de un cromosoma y, en ese contexto, no puede ser individualizado.
Para el aislamiento de un gen o de fragmentos más pequeños, el
DNA debe ser fragmentado. Si bien la ruptura del DNA puede ser realizada mecánicamente,
por este medio la fragmentación se produce al azar. La obtención
de fragmentos específicos fue posible mediante un método desarrollado
a partir de herramientas propias de ciertos organismos.
Para avanzar hacia un estudio
más detallado del DNA fue necesaria una metodología para obtener
grandes cantidades de fragmentos específicos de DNA. Estos fragmentos
podían ser DNA genómico, cDNA o DNA obtenidos a partir de oligonucleótidos
sintéticos.
A menudo, antes de que un determinado
fragmento de DNA o de mRNA pueda ser manipulado de cualquier modo, debe primero
ser localizado. Los cromosomas, incluso los de las células eucarióticas
más simples, contienen una enorme cantidad de DNA, por lo que localizar
un segmento específico es como tratar de encontrar la proverbial aguja
en el pajar. Para localizar fragmentos específicos se utiliza la técnica
de hibridación de ácidos nucleicos.
Con el desarrollo de técnicas
para cortar moléculas de DNA y multiplicar los fragmentos, hoy es posible,
en principio, determinar la secuencia de nucleótidos de cualquier fragmento
de ácido nucleico aislado. Para secuenciar una molécula de DNA
de gran tamaño, como el genoma completo de un virus, es preciso analizar
porciones pequeñas y posteriormente integrar los resultados.
En los comienzos de la investigación
del DNA recombinante, los biólogos se dieron cuenta de que los segmentos
de DNA que codifican ciertas proteínas (particularmente las de importancia
médica o agrícola) pueden transferirse a bacterias y ser expresados.
Las bacterias pueden funcionar como "fábricas" que suministran
una fuente virtualmente ilimitada de proteínas. Esta propiedad de las
bacterias fue aprovechada por los científicos y así nació
la biotecnología.
La metodología del DNA
recombinante, permite transferir genes tanto a células procarióticas
como a células vegetales y a otros organismos eucariotas.
Aislamiento de fragmentos específicos
de DNA
Los fragmentos específicos
de DNA se pueden obtener cortando moléculas de DNA con enzimas de restricción,
transcribiendo el mRNA a DNA con la enzima transcriptasa inversa, o por medio
de la síntesis de oligonucleótidos en el laboratorio. Las enzimas
de restricción se encuentran en la naturaleza en las células bacterianas
y en algunos bacteriófagos. Escinden las moléculas de DNA en secuencias
de reconocimiento específicas, que típicamente tienen de 4 a 8
nucleótidos de longitud. Su función en las células bacterianas
es degradar moléculas de DNA extrañas. El DNA de la bacteria se
protege de sus propias enzimas de restricción por la metilación
de nucleótidos en las secuencias de reconocimiento.
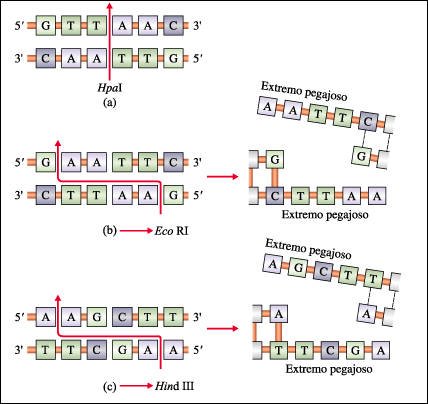
Algunas enzimas de restricción producen cortes de la molécula de DNA que dejan extremos rectos. Otras cortan de manera escalonada, dejando extremos "pegajosos" que luego pueden unirse, por apareamiento de bases complementarias, con otros fragmentos producidos por la misma enzima. Esto hace posible combinar segmentos de DNA de fuentes diferentes.. Una misma molécula de DNA producirá distintos fragmentos, llamados fragmentos de restricción, si es tratada o digerida con diferentes enzimas de restricción. El gran número de enzimas de restricción y de secuencias de reconocimiento diferentes hace posible que se las utilice para empalmar segmentos de DNA genómico de una variedad ilimitada de fuentes.
Las secuencias de nucleótidos de DNA reconocidas por tres enzimas de restricción ampliamente usadas: a) HpaI, b) EcoRI y c) HindIII.
Las secuencias de reconocimiento
de las enzimas de restricción frecuentemente tienen seis pares de bases
de longitud y, cuando se leen en la misma dirección (por ejemplo 5' a
3'), las dos cadenas complementarias de la secuencia son idénticas; estas
secuencias se denominan secuencias palindrómicas. Algunas enzimas como
EcoRI y HindIII escinden el DNA dando como resultado extremos "pegajosos".
Las enzimas de restricción generalmente se obtienen de bacterias y su
nombre deriva del nombre científico de esas bacterias: HpaI es de Hemophilus
parainfluenzae; EcoRI es de E. coli y HindIII es de Hemophilus influenzae.
Otra herramienta para obtener
fragmentos específicos de DNA la transcriptasa inversa que fue aislada
de ciertos virus que contienen genoma de RNA, los retrovirus. Esta enzima es
capaz de sintetizar DNA a partir de un molde de mRNA.
Cuando estos virus infectan una célula hospedadora, la transcriptasa inversa cataliza la síntesis de DNA a partir del molde de RNA viral; el mRNA viral, que codifica proteínas virales, se transcribe a partir de este DNA, como también el RNA viral que será empaquetado en nuevas partículas virales. En el laboratorio, la transcriptasa inversa puede utilizarse para sintetizar DNA a partir de un molde de RNA, segmentos que se conocen como DNA complementario, o cDNA.
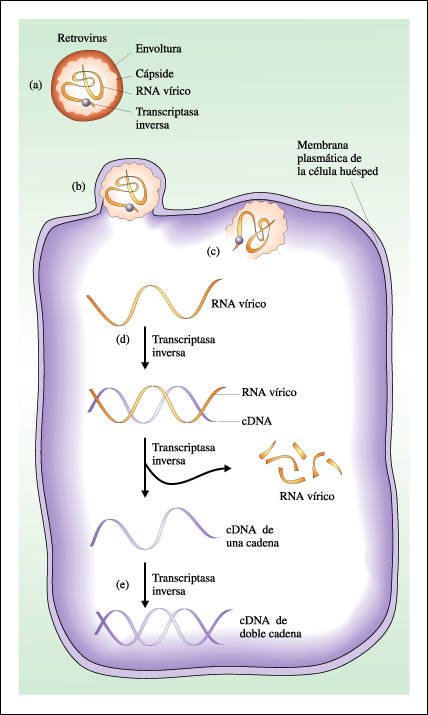
Infección de una célula animal por un retrovirus.
En el esquema anterior se observa
que
a. la cápside de un retrovirus
está rodeada típicamente por una envoltura externa de lipoproteína
formada por elementos de la membrana celular de su hospedador anterior y por
proteínas virales. Esta envoltura puede fusionarse con la membrana celular
de un nuevo hospedador, permitiendo que el virus entre en la célula.
b. Una vez que el retrovirus
ha entrado en la célula, el RNA viral se libera de la cápside
y
c. se transcribe a una única
cadena de DNA complementaria (cDNA).
d. Comienza de inmediato la
síntesis de la segunda cadena de DNA (complementaria a la primera), produciéndose
una molécula de cDNA de doblecadena.
Estas reacciones, así como la de degradación de la molécula
original del RNA viral, son catalizadas por la enzima transcriptasa inversa.
El cDNA de doble cadena puede
integrarse al cromosoma de la célula hospedadora. Posteriormente se transcriben,
a partir del DNA viral integrado, tanto el nuevo RNA genómico viral como
el mRNA para la síntesis de proteínas virales.
Para separar fragmentos de DNA
menores de 500 nucleótidos se utiliza como matriz un gel de poliacrilamida.El
tamaño de poro de este gel permite separar moléculas cuyo tamaño
difiere en un solo nucleótido. Para separar moléculas de DNA de
mayor tamaño, se utilizan matrices con poros mayores, como los geles
de agarosa, un polisacárido aislado de ciertas algas.
Los fragmentos de DNA separados
no pueden ser visualizados directamente, por ejemplo, mediante el uso de técnicas
de tinción.
Obtención de múltiples
copias de DNA
La maquinaria para duplicar
DNA ya estaba presente en la E. coli y en otras células bacterianas.
Pero hacían falta vectores que pudiesen llevar las moléculas de
DNA de interés al interior de estas células e iniciar la replicación.
Nuevamente, los procariotas y los virus aportaron la solución.
Los clones, en genética
molecular, son copias múltiples de la misma secuencia de DNA. Las secuencias
a ser clonadas se introducen en células bacterianas por medio de vectores.
Los plásmidos y los bacteriófagos se usan como vectores; los cósmidos
son vectores sintéticos que combinan características del fago
lambda (que tienen los extremos cohesivos o regiones COS) con propiedades de
los plásmidos. La secuencias de DNA que se desean clonar deben ser insertadas
en los vectores. Una vez en la bacteria, el vector y el DNA extraño que
lleva se replican y las copias múltiples pueden ser recuperadas de las
células.
Para clonar genes específicos se puede preparar una biblioteca genómica que consiste en una colección de fragmentos de DNA genómicos, clonados en vectores (plásmidos, bacteriófagos o cósmidos), que representan el genoma entero del organismo. Otra alternativa son las bibliotecas de cDNA que representan la colección completa de los mRNA que se sintetizan en un momento en una célula dada.
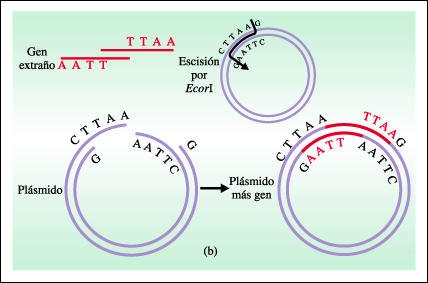
La enzima de restricción EcoRI escinde este plásmido en la secuencia GAATTC, dejando extremos "pegajosos" expuestos.
Los extremos pegajosos, formados
por secuencias TTAA y AATT, pueden unirse con cualquier otro segmento de DNA
que haya sido escindido por la misma enzima. Así es posible insertar
un gen extraño en el plásmido. (En este diagrama, la longitud
de las secuencias GAATTC se ha exagerado y las longitudes de las otras porciones
del gen extraño y del plásmido se han acortado). Cuando estos
plásmidos recombinantes, que llevan un gen extraño, se incuban
en ciertas condiciones con bacterias en medio líquido, son captados por
algunas de las células bacterianas. Cuando estas células se multiplican,
los plásmidos también se replican; el resultado es un número
creciente de células, todas sintetizando copias del mismo plásmido.
Los plásmidos pueden separarse luego de los otros contenidos celulares
y tratarse con EcoRI para liberar copias múltiples del gen clonado
Hasta la década de 1980, el único método para obtener grandes cantidades de un fragmento de DNA era clonándolo en vectores adecuados e introduciéndolo y multiplicándolo en bacterias. En el año 1986, un investigador norteamericano, K. Mullis, desarrolló un método que permite, a partir de una muestra muy pequeña de DNA, obtener millones de copias de DNA in vitro, en unas pocas horas y sin necesidad de usar células vivas. Esta técnica, llamada reacción en cadena de la polimerasa (PCR), requiere conocer la secuencia de nucleótidos de los extremos del fragmento que se quiere amplificar. Estas secuencias se usan para diseñar dos oligonucleótidos sintéticos de DNA complementarios a una porción de cada una de las dos cadena de la doble hélice.
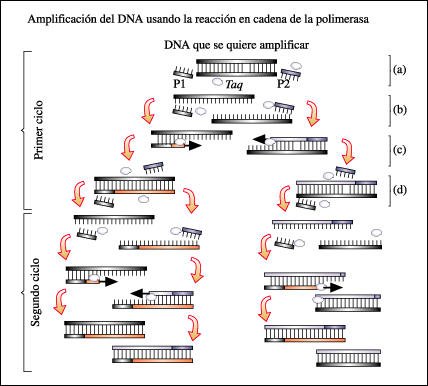
Reacción en cadena de la polimerasa (PCR).
a. La mezcla de reacción contiene la secuencia de DNA que se quiere amplificar,
dos oligonucleótidos sintéticos (P1 y P2) que servirán
como cebadores, una DNA polimerasa termoestable (Taq) y los cuatro desoxirribonucleótidos
trifosfato -dATP, dGTP, dCTP y dTTP-.
b. La mezcla de reacción
se somete a ciclos sucesivos, cada uno correspondiente a una fase de desnaturalización,
una de hibridación y una de elongación. Durante la desnaturalización,
que se realiza por calentamiento de la mezcla a 95 ºC, se separan las dos
cadenas del DNA molde.
c. Durante la hibridación,
la temperatura de incubación se reduce para permitir el apareamiento
de las bases de ambos cebadores en el sitio donde encuentran una secuencia complementaria.
d. Durante la fase de elongación,
la mezcla se calienta a 72 ºC, temperatura a la cual la DNA polimerasa
extiende la cadena complementaria a partir del extremo 3' de los cebadores.
Al finalizar cada ciclo, la cantidad de DNA molde disponible para el ciclo siguiente
aumenta al doble.
Entre muchas de las aplicaciones
que la PCR pone a disposición se encuentran la detección precoz
o prenatal de enfermedades genéticas, la detección de infecciones
virales latentes o la producción de grandes cantidades de fragmentos
de DNA humano a una velocidad muy superior a la posible mediante otros métodos.
Esta técnica también se aplica para estudios de identidad y filiación.
Localización de fragmentos
específicos de DNA: hibridación
Un fragmento de interés se puede identificar mediante técnicas de hibridación de ácidos nucleicos que dependen de la capacidad de una cadena simple de RNA o de DNA (que puede ser separada de la doble hélice por calentamiento) para combinarse o hibridar con otra cadena que tiene una secuencia de nucleótidos complementaria. Cuanto mayor sea la similitud entre las secuencias de nucleótidos de las dos cadenas, más rápida y más completa será la hibridación.
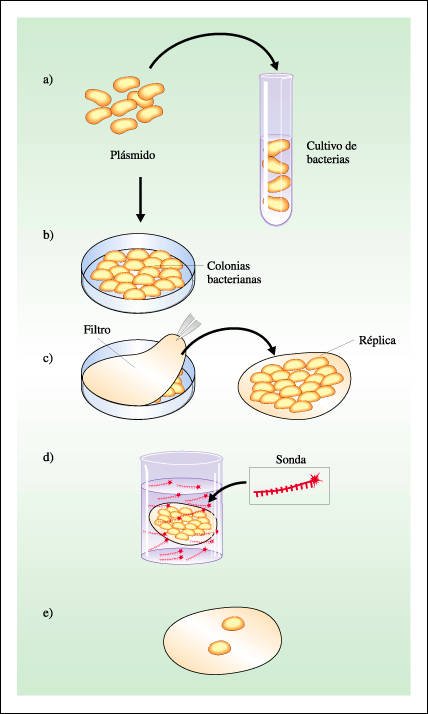
Transformación de bacterias y localización de un segmento de DNA de interés por medio de una sonda radiactiva
a. Las bacterias competentes
se transforman con plásmidos que contienen distintos fragmentos de DNA
obtenidos por digestión de un DNA genómico con una enzima de restricción.
Los plásmidos contienen, además, un gen de resistencia a un antibiótico
que se usará para la selección de las colonias que hayan incorporado
el plásmido.
b. Las bacterias transformadas
son distribuidas en una placa de Petri con un medio de cultivo sólido
que contiene el antibiótico de selección. Sólo las bacterias
que hayan adquirido el plásmido recombinante crecerán formando
colonias aisladas. Cada colonia se origina por divisiones sucesivas de una única
bacteria inicial y todas las células de una colonia contienen el mismo
plásmido recombinante.
c. Algunas bacterias de cada
colonia son transferidas (blotting) a un filtro especial obteniéndose
una réplica. La réplica es tratada con una solución de
alto pH para romper las células y desnaturalizar el DNA del plásmido.
El DNA se une químicamente al filtro
d. El filtro con el DNA desnaturalizado
es incubado en una solución que contiene una sonda radioactiva que consiste
en una molécula de DNA o RNA de cadena simple, complementaria al segmento
de DNA de interés. Se permite que la sonda se hibride.
e. Se lava del filtro el exceso
de solución que contiene las sondas que no han hibridado. Las moléculas
de doble cadena que se hayan formado permanecen en su lugar en el filtro y su
visualización se realiza por medio de autorradiografía, técnica
en la cual primero, cada filtro es cubierto con una película fotográfica
y dejado en la oscuridad. Durante el período de exposición, el
radioisótopo libera energía que impacta la emulsión fotográfica.
Luego del revelado de la película, la posición del fragmento radioactivo
se observa como una marca oscura, producida por un depósito de plata
de la emulsión. Las réplicas de las colonias que estén
marcadas con la sonda radiactiva después de este tratamiento, identifican
las colonias originales que contenían vectores con el segmento de DNA
que se deseaba estudiar.
Existe también una técnica
que permite localizar secuencias específicas de DNA o RNA en células
y tejidos mediante el uso de sondas de ácidos nucleicos. Esta técnica,
conocida como hibridación in situ, permite, por ejemplo, localizar una
determinada secuencia en un cromosoma. Las sondas, que también pueden
ser marcadas con un colorante fluorescente, son hibridizadas a los cromosomas,
expuestos previamente a un alto pH que rompe las uniones puentes de hidrógeno
y separa las dos cadenas de DNA.
El método de hibridación in situ también puede ser usado para detectar la presencia de moléculas de RNA específicas en células de distintos tejidos e indicar, de esta manera la expresión diferencial de genes en esos tejidos.
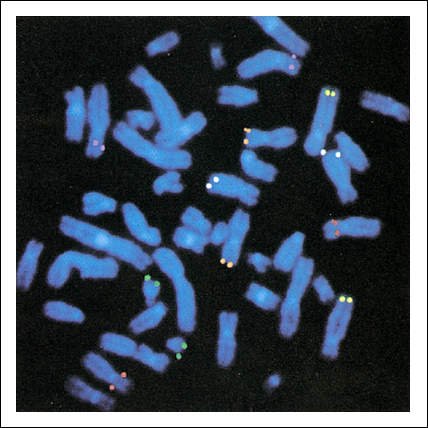
Hibridación in situ. Cromosomas humanos en metafase, en los cuales se identifican secuencias específicas de nucleótidos mediante la técnica de hibridación in situ de ácidos nucleicos.
En la hibridación in
situ, las sondas están marcadas con colorantes fluorescentes. Cada color
indica la localización de una secuencia de DNA diferente: rojo, localización
en el cromosoma X del gen de la distrofia muscular infantil mas común;
verde, región del cromosoma 21 donde se encuentra el gen responsable
del Síndrome de Down; blanco, un oncogen en el cromosoma 8; violeta,
un gen que codifica una proteína de membrana de los glóbulos blancos;
amarillo, una secuencia sin determinar en el cromosoma 5.
En los últimos años,
se ha desarrollado una técnica que promete generar un salto cualitativo
en los estudios de la expresión genética. Se trata de los microarreglos
o chips de DNA que permiten monitorear la expresión de un genoma en forma
integrada y en un tiempo récord.
La base de la técnica
es simplemente la hibridación de secuencias complementarias de ácidos
nucleicos a una escala microscópica. Este altísimo nivel de miniaturización
permite que la cantidad de sonda requerida sea mínima Con el empleo de
diferentes "etiquetas" fluorescentes se pueden comparar los patrones
de hibridación de dos tipos celulares cualesquiera encontrando rápidamente
aquellos genes que poseen un patrón de expresión diferencial.
De este modo es posible detectar genes específicamente activados en células
que han sufrido algún tipo de diferenciación, como células
de diferentes tejidos, tumorales, sometidas a ciertos tratamientos, etc.
Los microchips proveen una revolucionaria
herramienta para explorar la expresión génica y el análisis
de secuencias tanto en la investigación básica como aplicada.
Secuenciación del DNA
La secuenciación del
DNA es la determinación de la secuencia de nucleótidos de una
molécula de DNA. Se usan dos técnicas principales de secuenciación:
una implica métodos enzimáticos y la otra métodos químicos.
La secuenciación depende
de la disponibilidad de copias múltiples de un fragmento de DNA, obtenidas
por clonado de un segmento producido por digestión con enzimas de restricción
o por la técnica de PCR. Esta técnica se vale de oligonucleótidos
sintéticos y de una DNA polimerasa termoestable.
Combinando la información
de la secuenciación de distintos fragmentos del mismo DNA producidos
por diferentes enzimas de restricción, los biólogos moleculares
pueden determinar la secuencia completa de un segmento largo de DNA (como por
ejemplo un gen entero).
Existen dos métodos de
secuenciación. En la secuenciación de un segmento de una molécula
de DNA por el método de Maxam y Gilbert, el segmento de cadena simple
(presente en múltiples copias) se marca radiactivamente en el extremo
5'. La solución que contiene al DNA marcado se divide en cuatro porciones,
cada una de las cuales se somete a un tratamiento químico diferente para
romper las moléculas en una sola de las cuatro bases nitrogenedas. Con
los fragmentos resultantes se realiza una electroforesis en un gel de poliacrilamida
desnaturalizante, en el que se separan fragmentos que difieren incluso en un
nucleótido de longitud. Combinando la información obtenida con
cada reacción, se puede inferir la secuencia del segmento completo.
En la secuenciación por
el método enzimático, se procede en varias etapas. En una primera
etapa, el oligonucleótido iniciador marcado radiactivamente se aparea
con el DNA de simple cadena a secuenciar y se inicia la síntesis de la
cadena complementaria por parte de la DNA polimerasa. La mezcla en la que se
producirá la reacción de síntesis se separa en 4 tubos,
cada uno de los cuales contiene, además, uno de los nucleótidos
terminadores (ddATP, ddCTP, ddGTP o ddTTP). Estos son dideoxinucleótidos
que carecen del OH- en la posición 3', por lo que, una vez que son agregados
a la cadena que se está sintetizando, no pueden reaccionar con ningún
otro nucleótido y se transforman, así, en el último nucleótido
de la cadena.
En una segunda etapa, los productos
de reacción son sembrados en la parte superior de un gel, en "calles"
separadas. Luego de la corrida electroforética, se realiza la autorradiografía
del gel que permite leer la secuencia complementaria del DNA original. En el
recuadro que se encuentra a la izquierda de la fotografía del gel, que
esquematiza la cuba electroforética, se señalan las posiciones
de los fragmentos de DNA de una porción aumentada del gel. Las posiciones
de los fragmentos de DNA permiten leer la secuencia de nucleótidos incógnita
de la sigueiente manera: por ejemplo, si el un fragmento de un solo nucleótido,
que se ubica en la primera posición en la calle correspondiente a la
guanina indica que el primer nucléotido de la secuencia contiene, como
base nitrogenada, una guanina.
De la misma manera, si buscamos
en qué calle se ubica el fragmento de dos nucleótidos, verificaremos
que el segundo nucléotido de la secuencia contiene, como base nitrogenada,
también una guanina. De esta manera, se puede deducir, a partir de la
ubicación de los fragmentos en el gel, la secuencia de nucleótidos
complementarios de la cadena incógnita. La secuencia completa de nucleótidos
se presenta a la derecha del recuadro que esquematiza el gel y es: 5' GGACAATTGT
3'.
La secuenciación depende
de la disponibilidad de copias múltiples de un fragmento de DNA, obtenidas
por clonado de un segmento producido por digestión con enzimas de restricción
o por la técnica de PCR. Esta técnica se vale de oligonucleótidos
sintéticos y de una DNA polimerasa termoestable.
Combinando la información
de la secuenciación de distintos fragmentos del mismo DNA producidos
por diferentes enzimas de restricción, los biólogos moleculares
pueden determinar la secuencia completa de un segmento largo de DNA (como por
ejemplo un gen entero).
Biotecnología
El término biotecnología
fue creado en 1917 por el ingeniero húngaro Karl Ereky para describir
procesos en los que se formaban productos a partir de materiales crudos, con
la ayuda de la actividad metabólica de organismos vivos. Hoy el término
biotecnología engloba todo tipo de producción industrial de "bienes
y servicios" por medio de procesos que utilizan organismos, sistemas o
procesos biológicos.
Las técnicas de DNA recombinante
permiten la construcción de vectores, portadores de genes específicos,
que son introducidos en células bacterianas, las cuales sintetizan las
proteínas codificadas por los genes.
La primera síntesis de
una proteína de mamífero en una célula bacteriana fue realizada
usando el gen para la hormona somatostatina, una proteína pequeña
de sólo 14 aminoácidos que podía ser detectada en cantidades
muy pequeñas.
Más recientemente, se
ha logrado introducir en células bacterianas genes para otras proteínas
útiles en medicina, que han funcionado correctamente para la síntesis
de las proteínas codificadas. Los plásmidos utilizados para la
expresión de proteínas foráneas se conocen con el nombre
de vectores de expresión. Un ejemplo es el gen para la insulina humana.
Otro es el gen para la somatotropina u hormona de crecimiento, que se utiliza
para tratar cierta forma de enanismo en los niños.
Desde el punto de vista económico,
la síntesis bacteriana de otras proteínas es de importancia creciente.
Por ejemplo, la enzima renina, que se extrae del estómago de terneros
y se usa en la industria láctea para elaborar queso, ha sido producida
por tecnología de DNA recombinante. Más recientementese ha logrado
inducir la síntesis bacteriana de la enzima celulasa, producida en la
naturaleza por ciertos hongos. Esta enzima convierte a la celulosa, que no es
digerible por la mayoría de los organismos, en glucosa, la molécula
alimenticia de gran importancia.
Las vacunas contra enfermedades virales son otro producto importante de la biotecnología. Todos los virus, como se sabe, consisten en ácido nucleico envuelto por una cubierta proteínica. Son las proteínas exteriores del virus las que determinan si el virus puede unirse o no a la célula blanco y penetrar en ella. En el torrente sanguíneo de los animales, estas proteínas del virus, reconocidas como extrañas por células del sistema inmune, generan la formación de anticuerpos, moléculas que desempeñan un papel central en la inmunidad futura contra el virus. La mayoría de las vacunas se hacen utilizando partículas virales inactivas o modificadas. Las vacunas producidas exclusivamente a partir de las proteínas virales externas sintetizadas en bacterias son más seguras dado que, sin el ácido nucleico viral, no puede ocurrir contaminación de la vacuna por partículas infectivas.
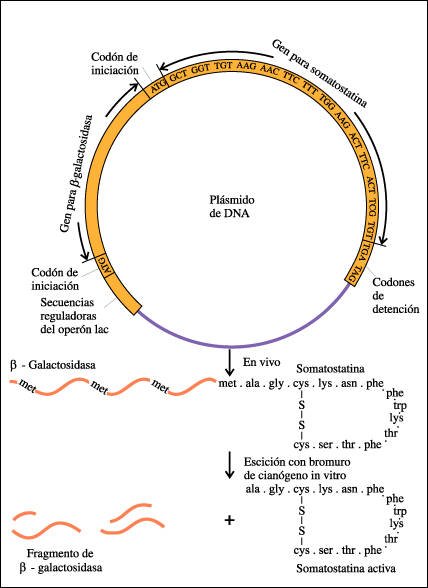
El proyecto de la somatostatina.
Un gen para somatostatina sintetizado artificialmente se fusiona con el gen
para la beta-galactosidasa de un plásmido bacteriano. Introducido en
E. coli, este plásmido dirige la síntesis de una proteína
híbrida, que comienza como betagalactosidasa, pero termina como somatostatina.
El bromuro de cianógeno escinde la proteína en el sitio de la
metionina, liberando así la hormona intacta, junto con muchos fragmentos
de beta-galactosidasa, de los que puede ser separada. Leyendo en el sentido
del movimiento de las agujas del reloj, la secuencia de nucleótidos que
se muestra en este diagrama es la secuencia 5' a 3' de la cadena inactiva de
la doble hélice de DNA. Es complementaria de la cadena molde a partir
de la cual se transcribe el mRNA y, con excepción de la sustitución
de la timina por uracilo, es idéntica a la secuencia 5' a 3' de la molécula
de mRNA transcripta.
Transferencia de genes
La metodología del DNA
recombinante, que permite la modificación de plásmidos para transferir
genes tanto a células procarióticas como a células vegetales,
se utiliza para desarrollar vectores adecuados para la transferencia de genes
a otros organismos eucarióticos.
Uno de los problemas técnicos con que se tropieza cuando se intenta transferir
genes, es saber si un gen determinado realmente ha sido introducido en una nueva
célula hospedadora y, si una vez transferido, está dirigiendo
la síntesis de proteína.

Agallas de corona creciendo sobre un tallo de tabaco.
El Agrobacterium tumefacienses
una bacteria común del suelo que infecta a las plantas, produciendo una
tumefacción o tumor del tejido, conocido como agalla de corona. Las investigaciones
han mostrado que la causa de esta agalla no es el A. tumefaciens mismo, sino
un plásmido relativamente grande contenido en la bacteria. Parte de este
plásmido, que se conoce como Ti (inductor de tumor), se integra al DNA
de la célula vegetal hospedadora.
Para tratar de comprender de qué manera el plásmido Ti ejerce sus efectos, se ha usado la tecnología de DNA recombinante para examinar sus genes. Tres de sus genes, según se ha encontrado, rigen la síntesis de hormonas vegetales, que actúan directamente sobre las células de la agalla para promover su crecimiento. Uno o más genes adicionales subvierten la maquinaria celular para producir aminoácidos particulares, llamados opinas, que pueden ser utilizados por las células de la agalla, pero no por las células normales. Además, las opinas actúan, de alguna manera, como "afrodisíacos moleculares", incrementando la conjugación bacteriana y promoviendo así la diseminación del plásmido Ti en bacterias no infectadas. En efecto, el Ti asume y dirige las actividades de sus dos hospedadores, las células bacterianas y las células vegetales, para promover su propia multiplicación. El plásmido Ti ha suscitado considerable atención no sólo a raíz de sus notables propiedades, sino también por ser un vector potencial para transportar genes útiles a plantas cultivadas.

Un caso interesante de transferencia de genes se ilustra en este notable experimento en el que aislaron el gen para la enzima luciferasa de las luciérnagas.
El sustrato para la enzima luciferasa
es una proteína llamada luciferina. En presencia de oxígeno, la
luciferina junto con luciferasa y ATP producen bioluminiscencia, como se ve
en el destello de la luciérnaga. El gen de la luciferasa fue clonado
en E. coli y luego empalmado en el cromosoma de un virus vegetal, lo cual le
suministró una secuencia regulatoria. El cromosoma viral modificado se
insertó luego en plásmidos Ti, los plásmidos fueron transferidos
a las bacterias y las bacterias se incubaron con células foliares del
tabaco. Las células formaron una masa de tejido, conocido como callo,
a partir del cual se obtuvieron nuevas plantas en medio de crecimiento adecuado.
Las nuevas plantas fueron regadas con una solución que contenía
luciferina y al cabo de un tiempo las plantas resplandecían.


